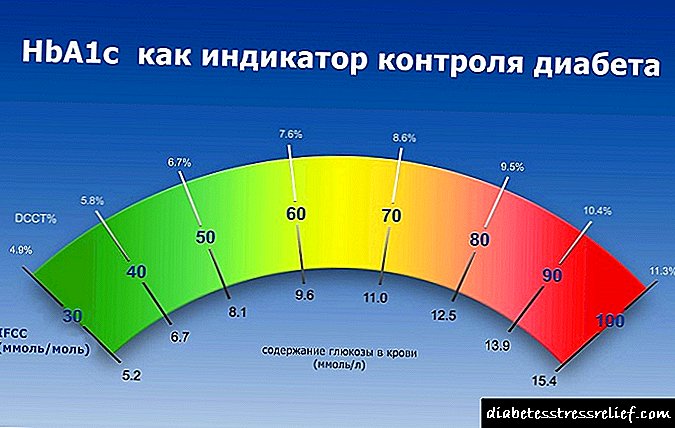
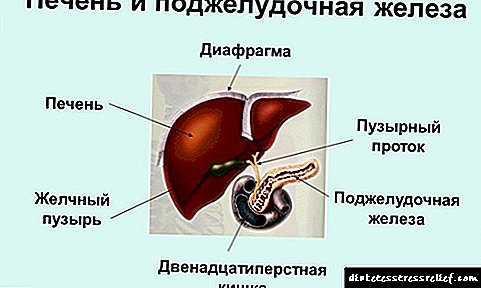
Ketonuria គឺជាវត្តមាននៅក្នុងទឹកនោមរបស់សាកសព ketone គឺអាសេតូន
យន្តការនៃ ketonuria ។
នៅក្នុងមនុស្សដែលមានសុខភាពល្អកាបូអ៊ីដ្រាតខ្លាញ់និងប្រូតេអ៊ីនមួយចំនួនត្រូវបានកត់សុីទៅនឹងកាបូនឌីអុកស៊ីតនិងទឹក។ នៅក្នុងស្ថានភាពរោគសាស្ត្រមួយចំនួនជាពិសេសជំងឺទឹកនោមផ្អែមការផលិតអាំងស៊ុយលីនមានការថយចុះ។ នៅក្នុងថ្លើមបំរុងគ្លីកូហ្សែនត្រូវបានកាត់បន្ថយជាលិការាងកាយជាច្រើនស្ថិតនៅក្នុងស្ថានភាពនៃភាពអត់ឃ្លានថាមពល។ នៅក្រោមលក្ខខណ្ឌទាំងនេះដំណើរការនៃការកត់សុីនៃប្រូតេអ៊ីននិងជាតិខ្លាញ់ក្នុងថ្លើមត្រូវបានធ្វើឱ្យសកម្មប៉ុន្តែកង្វះ glycogen នាំឱ្យមានការកត់សុីមិនពេញលេញនិងការប្រមូលផ្តុំនៅក្នុងឈាមនៃផលិតផលដែលមិនមានអុកស៊ីតកម្មនៃ lipid និងការរំលាយអាហារប្រូតេអ៊ីន - រាងកាយ ketone ។ ដោយសារតែការប្រមូលផ្តុំនៃសាកសព ketone នៅក្នុងឈាម (ketonemia), pH នៃឈាមផ្លាស់ប្តូរទៅផ្នែកខាងអាស៊ីត។ លក្ខខណ្ឌនេះត្រូវបានគេហៅថាទឹកអាស៊ីត។ ទឹកនោមរបស់អ្នកជំងឺបែបនេះមានប្រតិកម្មអាស៊ីដខ្លាំងនិងក្លិនអាសេតូន។
ភាពអត់ឃ្លានការប្រើប្រាស់អាហារប្រូតេអ៊ីននិងខ្លាញ់ជាចម្បងការមិនរាប់បញ្ចូលកាបូអ៊ីដ្រាតពីរបបអាហារនាំឱ្យមានការកើនឡើងនៃការបង្កើតរាងកាយរបស់ ketone និងការបញ្ចេញទឹកនោមរបស់ពួកគេ។
នៅវ័យក្មេង ketonuria មានលក្ខណៈទូទៅជាងមនុស្សពេញវ័យហើយមិនមានសារៈសំខាន់ព្យាបាលទេ។ បាតុភូតនេះគួរឱ្យចាប់អារម្មណ៍ចំពោះគ្រូពេទ្យកុមារជាមួយ "ក្អួតអាសេទិក" ដែលត្រូវបានផ្សារភ្ជាប់ជាមួយនឹងកំហុសក្នុងរបបអាហារ។
Ketonuria នៅក្នុងរយៈពេលក្រោយការវះកាត់គឺដោយសារតែការបំបែកប្រូតេអ៊ីនដោយសារតែការប៉ះទង្គិចវះកាត់។
ចំពោះមនុស្សវ័យចំណាស់ ketonuria កើតឡើងក្នុងទម្រង់ធ្ងន់ធ្ងរនៃជំងឺទឹកនោមផ្អែមហើយមានតម្លៃខ្ពស់ក្នុងការធ្វើរោគវិនិច្ឆ័យ។ ចំះកុមារវាអាចមានជំងឺផ្សង ៗ យសារការធ្វើចលនារំលាយអាហារកាបូអ៊ីដ្រាត។ ហេតុដូច្នេះសូម្បីតែកំហុសតូចតាចនៅក្នុងរបបអាហារជាពិសេសនៅក្នុងវត្តមាននៃការឆ្លងមេរោគស្រួចស្រាវការរំភើបសរសៃប្រសាទការធ្វើការងារហួសប្រមាណជាដើម។ អាចនាំឱ្យមាន ketosis ។ Ketonuria ក្នុងវ័យកុមារភាពអាចត្រូវបានគេសង្កេតឃើញដោយជំងឺ toxicosis, ជំងឺក្រពះពោះវៀនយូរ, ជំងឺរាគរូសនិងជំងឺដទៃទៀត។ ចំពោះទារកទើបនឹងកើតការកើនឡើងនៃ ketones ទឹកនោមតែងតែបណ្តាលមកពីកង្វះអាហារូបត្ថម្ភ។ Ketonuria ដែលត្រូវបានគេសង្កេតឃើញនៅក្នុងជំងឺឆ្លង - គ្រុនក្តៅក្រហម, គ្រុនផ្តាសាយ, ជំងឺរលាកស្រោមខួរជួរនិងការស្រវឹងគឺជាសញ្ញាបន្ទាប់បន្សំ, ការឆ្លងនិងមិនមានតម្លៃខ្ពស់ក្នុងការធ្វើរោគវិនិច្ឆ័យ។
Ketonuria សម្រាប់ជំងឺទឹកនោមផ្អែម មានការរីកចម្រើនដោយសារតែការកើនឡើង ketogenesis និង ketolysis ខ្សោយ។ ការកើនឡើង ketogenesis នាំឱ្យមានការកើនឡើងនៃការប្រមូលផ្តុំខ្លាញ់ពីជាលិកា adipose ការថយចុះនៃការបង្កើត oxalacetate នៅក្នុងវដ្ត Krebs និងការថយចុះនៃជីវគីមីអាស៊ីតខ្លាញ់ថយចុះ។ នៅក្នុងជំងឺទឹកនោមផ្អែមធ្ងន់ធ្ងរអមដោយការបំផ្លាញជាលិកាតំរងនោម (កន្លែងសម្រាប់ការបំផ្លាញ ketones) ការរំលោភបន្ថែមនៃ ketolysis កើតឡើង។
អវត្ដមាននៃគ្លុយកូសនៅក្នុងវត្តមាននៃ ketonuria មិនរាប់បញ្ចូលជំងឺទឹកនោមផ្អែម។
ជាធម្មតាសាកសព ketone ត្រូវបានបង្កើតឡើងក្នុងចំនួនតិចតួចពីផលិតផលចុងក្រោយនៃការរំលាយអាហារកាបូអ៊ីដ្រាតនិងខ្លាញ់ - អេទីល - កូអា។2-bodies) តាមរយៈអាសុីកូកាលីឡី - កូនិងត្រូវបានបន្សាបស្ទើរតែទាំងស្រុង។
នៅក្នុងជំងឺទឹកនោមផ្អែមការប្រមូលផ្តុំជាតិខ្លាញ់កើនឡើងជាមួយនឹងការបង្កើតបរិមាណអាសេទីលកូកា (C2រាងកាយ) ។ ក្នុងពេលតែមួយដោយសារតែការរំលោភលើការរំលាយអាហារកាបូអ៊ីដ្រាតការថយចុះនៃការបង្កើត oxalacetate ដែល C2ឧបករណ៍ត្រូវបានរួមបញ្ចូលនៅក្នុងវដ្ត Krebs និងដុតរហូតដល់កាបូនឌីអុកស៊ីតនិងទឹក។ លើសពីនេះទៀតជាលទ្ធផលនៃការកើនឡើងនៃការបំបែកជាតិខ្លាញ់ការបំលែងជីវគីមីបញ្ច្រាសនៃ C ត្រូវបានរារាំង។2រាងកាយទៅជាអាស៊ីតខ្លាញ់។ ដូច្នេះបរិមាណដ៏ច្រើននៃកកកុញ C2- អ្នកដែលនាំទៅរកការផលិតកូកាដ៏ច្រើនហើយជាលទ្ធផលអាសេតូនអាស៊ីតអាសេទិកនិងអាស៊ីតបេតាហាយដ្រូអ៊ីទ្រីកដែលត្រូវបានបញ្ចេញនៅក្នុងទឹកនោម។
សាកសពកេតតុន
ទាំងនេះគឺជាផលិតផលបំបែកមធ្យមដែលត្រូវបានបង្កើតឡើងក្នុងកំឡុងពេលបំពេញមុខងារថ្លើម។ ជាធម្មតាការរំលាយអាហារបណ្តេញសាកសពរបស់ ketone ទៅនឹងការរិចរិលថែមទៀត។
ទុនបម្រុងគ្លុយកូសរបស់រាងកាយត្រូវបានរកឃើញនៅក្នុងខ្លាញ់ក្នុងខ្លួនដែលជាហេតុធ្វើឱ្យ ketonuria គឺជាកង្វះជាតិកាបូអ៊ីដ្រាតខ្លាំង។ សាកសពកេតតុនគឺជាអ្នកផ្តល់ថាមពលដ៏ល្អបំផុត។ នៅក្នុងនេះសូម្បីតែអាស៊ីតខ្លាញ់ក៏នៅពីក្រោយពួកគេដែរ។ ដូច្នេះនៅពេលដែលខួរក្បាលឬបេះដូងខ្វះអាស៊ីតផូស្វ័ររាងកាយនឹងផលិតសាកសព ketone ភ្លាមៗក្នុងល្បឿនមួយ។
តើមានមូលហេតុអ្វីខ្លះ
Ketonuria គឺជាមាតិកាទឹកនោមរបស់សាកសព ketone ខ្ពស់ជាងធម្មតា។
នៅក្នុងរាងកាយមនុស្សមានសុខភាពល្អការរំលាយអាហារមានតុល្យភាពល្អ។ តើអ្វីអាចបណ្តាលឱ្យមានជំងឺ ketonuria?

- ភាពលេចធ្លោនៃប្រូតេអ៊ីននិងខ្លាញ់នៅក្នុងរបបអាហារ។ ដោយសារតែការថយចុះកាបូអ៊ីដ្រាតនៅក្នុងអាហារកោសិកាខ្វះអាហារូបត្ថម្ភ។ ប្រឆាំងនឹងផ្ទៃខាងក្រោយនេះ ketonuria មានការរីកចម្រើន។ នេះគឺជាប្រតិកម្មនៃរាងកាយទៅនឹងអតុល្យភាពនៃរបបអាហារ។
- របបអាហារនិងការរំលោភបំពានភាពអត់ឃ្លានអាចបង្កឱ្យមានរូបរាងនៃសាកសពកេតតេននៅក្នុងទឹកនោម។ កំហុសក្នុងការជ្រើសរើសរបបអាហារនាំឱ្យមានការបំបែកខ្លាញ់យ៉ាងឆាប់រហ័សខណៈពេលដែលចំនួនអង់ស៊ីមកើនឡើង។ Ketonuria ក៏ជារូបរាងរបស់អាសេតូននៅក្នុងឈាមផងដែរ។ ជាធម្មតាប្រសិនបើមនុស្សម្នាក់ស្រេកឃ្លានអស់រយៈពេលជាង ៦ ថ្ងៃនោះមាតិកានៃសាកសព ketone នៅក្នុងខ្លួនមនុស្សកើនឡើងខ្ពស់ជាងធម្មតា។
- ការប្រើថ្នាំសន្លប់សម្រាប់ការវះកាត់។
- ជំងឺទឹកនោមផ្អែម។ ក្នុងករណីនេះ ketonuria លេចឡើងពីពេលមួយទៅពេលមួយ។ សាកសពកេតតុនតែម្នាក់ឯងមិនមែនជាបុព្វហេតុនៃជំងឺនេះទេ។ នៅក្នុងជម្ងឺទឹកនោមផ្អែមវគ្គសិក្សាបន្ថែមនៃកាបូអ៊ីដ្រាតនិងអាំងស៊ុយលីនត្រូវបានចេញវេជ្ជបញ្ជា។
- ការខះជាតិទឹក។ វាកើតឡើងនៅពេលដែលរាងកាយឡើងកំដៅឬជាផលវិបាកនៃជំងឺទឹកនោមផ្អែម។
- ជំងឺថ្លើមឬជំងឺឆ្លងធ្ងន់ធ្ងរ (ឧទាហរណ៍ជំងឺរាគរូស) ។
- ការវិវត្តនៃដុំសាច់នៅក្នុងបំពង់រំលាយអាហារ។
- ការមិនស្រួលនៅក្នុងលំពែង។
- ការពុលដោយជាតិអាល់កុលឬដោយសមាសធាតុគីមីដូចជាផូស្វ័រសំណ។
- ការបំផ្លាញប្រព័ន្ធសរសៃប្រសាទកណ្តាលការរំភើបសរសៃប្រសាទ។
ចងចាំថាក្លិនអាសេតូននៅពេលដកដង្ហើមឬនោមគឺជាសញ្ញាមួយដែលត្រូវទៅជួបគ្រូពេទ្យ។ វាក៏ជាហេតុផលដើម្បីផ្លាស់ប្តូររបៀបរស់នៅនិងតុល្យភាពអាហារូបត្ថម្ភ។
យកចិត្តទុកដាក់ចំពោះកុមារ
គ្លីនិកត្រូវបានពិគ្រោះជាញឹកញាប់ប្រសិនបើកុមារមានក្អួតក្លិនអាសេតូន។ ឬក្លិនបែបនេះបានលេចឡើងនៅក្នុងទឹកនោម។ ហើយទោះបីជាវាជាសញ្ញានៃ ketonuria ក៏ដោយក៏វាមិនចាំបាច់ជារោគសញ្ញានៃជំងឺធ្ងន់ធ្ងរដែរ។

ជារឿយៗអ្វីគ្រប់យ៉ាងត្រូវបានពន្យល់ដោយប្រព័ន្ធរំលាយអាហារមិនល្អឥតខ្ចោះ។ មូលហេតុនៃ ketonuria ចំពោះកុមារគឺជាលក្ខខណ្ឌមួយដែលបរិមាណថាមពលដ៏សំខាន់ត្រូវបានចំណាយ។ វាច្រើនតែកើតឡើងនៅពេល៖
- ការរំជួលចិត្ត
- បង្កើនកម្លាំងកាយសម្បទា
- របបអាហារគ្មានតុល្យភាព
- ផ្តាសាយ
ការពិតគឺថារាងកាយរបស់កុមារមិនមានផ្ទុកគ្លីកូហ្សែនច្រើនដូច្នេះការបំបែកខ្លាញ់យ៉ាងសកម្មកើតឡើងហើយសញ្ញានៃ ketonuria ត្រូវបានគេសង្កេតឃើញ។
មានផ្ទៃពោះ
អស់រយៈពេល ៩ ខែនៃការមានផ្ទៃពោះស្ត្រីជារឿយៗត្រូវធ្វើតេស្តទឹកនោម។ នេះគឺជាការចាំបាច់ដើម្បីកុំឱ្យខកខានគម្លាតបន្តិចបន្តួចពីបទដ្ឋាននៅក្នុងរាងកាយសម្រាប់រយៈពេលទាំងមូលនៃកាយវិការ។ យ៉ាងណាមិញសរីរាង្គទាំងអស់មានបន្ទុកធំ។ តើសាកសពកេតតេនមានអ្វីខ្លះនៅក្នុងទឹកនោម?
ជាធម្មតាមាតិកាតូចៗរបស់ពួកគេគឺជាបទដ្ឋាន។ ការវិភាគសាមញ្ញបំផុតនឹងត្រូវឆ្លងកាត់ការសាកល្បងយ៉ាងឆាប់រហ័ស។ អ្នកត្រូវបន្ថយបន្ទះតេស្តិ៍ក្នុងទឹកនោម។
ការធ្វើតេស្តអវិជ្ជមានត្រូវបានគេចាត់ទុកថាធម្មតាក្នុងអំឡុងពេលមានផ្ទៃពោះឬចំនួននៃ ketones គឺតិចតួចបំផុត។ ប្រសិនបើតម្លៃតេស្តនេះមានចាប់ពី 15 ទៅ 160 មីលីក្រាម / dl - នេះជាមូលហេតុនៃការព្រួយបារម្ភ។
ក្នុងអំឡុងពេលមានផ្ទៃពោះ ketonuria គឺជារោគសញ្ញាគួរឱ្យព្រួយបារម្ភ។ វាលេចឡើងជាមួយនឹងការពុលឆាប់។ ការពុលនៃរាងកាយហើយហេតុដូច្នេះទារកដែលមានអាសេតូនធ្វើឱ្យស្មុគស្មាញដល់ដំណើរនៃការមានផ្ទៃពោះ។

ប្រសិនបើស្ត្រីមានផ្ទៃពោះមានការបរាជ័យអ័រម៉ូនធ្ងន់ធ្ងរនោះនាងប្រឈមនឹងគ្រោះថ្នាក់។
ការកើនឡើងកម្រិតនៃ ketones នៅក្នុងទឹកនោមរបស់ទារកទើបនឹងកើតគឺដោយសារតែការបំបៅមិនគ្រប់គ្រាន់ឬកំហុសអាហារូបត្ថម្ភ។
រោគសញ្ញារបស់ Ketonuria
ប្រសិនបើកម្រិតនៃ ketones នៅក្នុងខ្លួនបានកើនឡើងគួរឱ្យកត់សម្គាល់នោះ ketonuria នឹងបង្ហាញខ្លួនវា:
- ហត់នឿយនឿយហត់,
- ក្លិនអាសេតូនចេញពីមាត់
- ក្លិនទឹកនោមអាសេតូន
- ការវិភាគនឹងបង្ហាញពីកំរិតខ្ពស់នៃកោសិកាឈាមសនៅក្នុងឈាម
- តេស្តឈាមជីវគីមីនឹងបង្ហាញមាតិកាគ្លុយកូសទាប
ប្រសិនបើ ketonuria បង្កឱ្យមានការលោតចូលក្នុងឈាមរបស់អាសេតូនបន្ទាប់មកវិបត្តិអាសេតូនអាចនឹងកើតឡើង។
ការកើនឡើងយ៉ាងខ្លាំងនៃអាស៊ីតនៅក្នុងកោសិកាអាចធ្វើឱ្យខូចខាតដល់សរីរាង្គខាងក្នុងយ៉ាងធ្ងន់ធ្ងរ។ ក្នុងករណីនេះប្រតិកម្មការពារត្រូវបានចាប់ផ្តើម - ក្អួត។
រោគសញ្ញារបស់ ketonuria ចំពោះកុមារ៖
- ពាក្យបណ្តឹងនៃការឈឺពោះ
- ពាក្យបណ្តឹងឈឺក្បាល
- ក្មេងហត់នឿយអស់កំលាំង
- ពាក្យបណ្តឹងនៃការចង្អោរ
- ក្អួត
- បង្កើនសីតុណ្ហភាពដល់ ៣៩ អង្សាសេ
- ការបដិសេធអាហារ
- មានក្លិនដូចអាសេតូន
- ថ្លើមរីក
តើអាចរកឃើញ ketonuria បានទេ?
មានតែការវិភាគគីមីប៉ុណ្ណោះដែលអាចរកឃើញនូវការកើនឡើងនៃកម្រិតនៃ ketone នៅក្នុងខ្លួន។ មន្ទីរពិសោធន៍នឹងបង្កើតបទដ្ឋាននៃសាកសព ketone ដែលមាននៅក្នុងនោះ។
នៅក្នុងថាំពទ្យទំនើបគេរកឃើញ ketonuria៖
- តេស្តឡង់,
- ការធ្វើតេស្តស្របច្បាប់
- គំរូឡេសស្តាត
- គំរូនៃរ័ត្នរ៉ា
- ការធ្វើតេស្តលឿន

ពិតណាស់ការធ្វើតេស្តលឿនគឺជារឿងធម្មតាបំផុត។ សកម្មភាពរបស់ពួកគេគឺផ្អែកលើប្រតិកម្មគីមីលទ្ធផលនៃការដែលអាចមើលឃើញស្ទើរតែភ្លាមៗ។ អ្នកណាម្នាក់ត្រូវដាក់បន្ទះសាកល្បងក្នុងទឹកនោមឬទម្លាក់វានៅលើកុំព្យូទ័របន្ទះសាកល្បង។ ក្នុងករណីមានប្រតិកម្មវិជ្ជមានតេស្តប្រែជាពណ៌ស្វាយ។ ពន្លឺនៃពណ៌អនុញ្ញាតឱ្យអ្នកវិនិច្ឆ័យដោយមាត្រដ្ឋានពណ៌ពិសេសអំពីចំនួនដងនៃរាងកាយរបស់ ketone ត្រូវបានលើស។
ចំណាត់ថ្នាក់អន្តរជាតិនៃជំងឺ
ចំណាត់ថ្នាក់ស្ថិតិអន្ដរជាតិនៃជំងឺនិងបញ្ហាសុខភាពដែលពាក់ព័ន្ធគឺជាមគ្គុទ្ទេសក៍យោងដែលសម្ភារៈអាចត្រូវបានប្រៀបធៀបជាអន្តរជាតិ។ ដោយមានជំនួយរបស់វាវិធីសាស្រ្តវិធីសាស្ត្រផ្សេងៗត្រូវបានបញ្ចូលគ្នា។ ស្ថិតិជំងឺកំពុងត្រូវបានប្រមូលនិងចាត់ថ្នាក់។ រៀងរាល់ ១០ ឆ្នាំម្តង IBC ត្រូវបានពិនិត្យឡើងវិញដោយគណៈកម្មការរបស់អង្គការសុខភាពពិភពលោក។ ដើម្បីចាត់ថ្នាក់និងវិភាគម៉ាស់ទាំងមូលនៃលទ្ធផលដែលប្រមូលបានពីចំណុចនៃស្ថិតិវាត្រូវបានបកប្រែទៅជាលេខអក្សរក្រមលេខ។ នៅក្នុងការងារបែបនេះអាយឌីស៊ីអភិវឌ្ឍន៍ត្រូវបានប្រើ។ សព្វថ្ងៃនេះវាត្រូវគ្នាទៅនឹង ICD-10 ។ វាមាន ២២ ថ្នាក់ (ផ្នែក) ។
យោងទៅតាមថតអាយឌីអេស ketonuria មានលេខកូដ R82.4 ។
ការការពារនិងរបបអាហាររបស់ Ketonuria
សម្រាប់ការការពារជំងឺតេតាណូរីវាចាំបាច់៖
- បរិភោគត្រឹមត្រូវ
- ដឹកនាំរបៀបរស់នៅដែលមានសុខភាពល្អ៖
- តាមដែលអាចធ្វើទៅបានដើម្បីឱ្យមានខ្យល់អាកាសបរិសុទ្ធ
- ធ្វើលំហាត់ប្រាណល្មម
- កុំចាប់ផ្តើមជំងឺរ៉ាំរ៉ៃ។

នៅក្នុងជំងឺរ៉ាំរ៉ៃដូចជាជំងឺទឹកនោមផ្អែមវេជ្ជបណ្ឌិតពិគ្រោះជាប្រព័ន្ធ។ ពីពេលមួយទៅពេលមួយវាចាំបាច់ក្នុងការធ្វើតេស្តជាក់ស្តែង។
ប្រតិកម្មវិជ្ជមានបង្ហាញពីរោគសាស្ត្រនៅក្នុងខ្លួន។ យកចិត្តទុកដាក់លើសញ្ញា! ការអំពាវនាវជាបន្ទាន់ទៅកាន់គ្លីនីកនឹងធ្វើឱ្យរាងកាយវិលត្រឡប់ទៅរកសភាពធម្មតាវិញឆាប់រហ័ស។ ដោយមិនចាកចេញពីរបបអាហារដែលវេជ្ជបណ្ឌិតបានកំណត់អ្នកអាចបង្កើនល្បឿននៃការជាសះស្បើយឡើងវិញ។
គោលការណ៍របបអាហាររបស់ Ketonuria៖
- កុំបរិភោគអាហារដែលមានប្រូតេអ៊ីននិងខ្លាញ់ខ្ពស់។
- ការទទួលទានកាបូអ៊ីដ្រាតធម្មតា
- ផឹកដំណោះស្រាយទឹកនិងសូដាបន្ថែមទៀត (ជាមួយជំងឺទឹកនោមផ្អែម - អាំងស៊ុយលីន) ។
មុខម្ហូបគួរតែមានៈសាច់គោឆ្អិននិងទន្សាយស៊ុបបន្លែជាច្រើនប្រភេទត្រីមានជាតិខ្លាញ់ទាប។ ធញ្ញជាតិមានប្រយោជន៍ដោយគ្មានប៊ឺបន្លែនិងផ្លែឈើ។ វាត្រូវបានផ្ដល់អនុសាសន៍ឱ្យផឹកទឹកផ្លែឈើភេសជ្ជៈផ្លែឈើបន្ថែម។
ដកចេញពីម៉ឺនុយ៖
- សាច់ខ្លាញ់
- រដូវហឺរ
- ផ្អែម
- ផ្លែក្រូច
- ចេក
- ផ្សិត
- អាហាររហ័ស។
ផ្តោតលើការព្យាបាល
Ketonuria មិនត្រូវបានព្យាបាលជាជំងឺដាច់ដោយឡែកទេ។ នាងគ្រាន់តែជាផលវិបាកប៉ុណ្ណោះ។ វាចាំបាច់ក្នុងការដកមូលហេតុដែលបណ្តាលឱ្យវាកើតឡើង។ ទីមួយការប្រឡងពេញលេញគឺចាំបាច់។ ភាពត្រឹមត្រូវនៃការធ្វើរោគវិនិច្ឆ័យនិងការបង្កើតមូលហេតុនៃ ketonuria ធានាការព្យាបាលដោយជោគជ័យ។
វេជ្ជបណ្ឌិតផ្តល់ការណែនាំខ្លះៗ៖
- ប្រសិនបើអ្នកលើសទម្ងន់អ្នកគួរតែរៀបចំថ្ងៃតមអាហារពីមួយពេលទៅមួយពេល។
- ប្រសិនបើការវិភាគបានបង្ហាញពីការកើនឡើងនៃ ketones ទឹកនោមសូមទិញការធ្វើតេស្តដើម្បីប្រើវានៅផ្ទះ។

- ទឹកនោមគួរតែត្រូវបានប្រមូលសម្រាប់ការវិភាគមិនលើសពីបួនម៉ោងមុនពេលសម្រាល។
- កុមារអាចស្រវឹងជាមួយភេសជ្ជៈអាល់កាឡាំងរៀងរាល់ 10-15 នាទីជាផ្នែកតូចៗ។ ធ្យូងដែលធ្វើឱ្យសកម្ម, enterosgel នឹងជួយសម្អាតបំពង់រំលាយអាហារ។
- ជាមួយនឹងការក្អួតនាពេលខាងមុខនេះវាមានប្រយោជន៍ក្នុងការផឹកភេសជ្ជៈប្រភាគ។ ហៅឡានពេទ្យ។
វិធីព្យាបាល ketonuria មានការណែនាំក្នុងវេជ្ជសាស្ត្រប្រជាប្រិយ។
- ជាមួយនឹងការក្អួតញឹកញាប់ផឹកទឹកសារធាតុរ៉ែទឹកផ្លែឈើស្ងួតដំណោះស្រាយគ្លុយកូស។ មួយស្លាបព្រាក្នុងរយៈពេលដប់នាទី។
- ដាក់លាមកមួយនៅផ្ទះ។ ដំបូងទឹកនៅសីតុណ្ហភាពបន្ទប់ក្រោយមកក្តៅចូលក្នុងសូដាមួយស្លាបព្រាកាហ្វេត្រូវបានបន្ថែម។
- ផឹក: រំលាយ 2 tbsp ក្នុង 1 លីត្រទឹក។ ទឹកឃ្មុំ, ចាក់ទឹកមួយ lemon ។ ផឹក 1 tbsp ។ រៀងរាល់ ១៥ នាទីម្តង។
- រូបមន្តសម្រាប់ដំណោះស្រាយសូដាៈរំលាយសូដា 1 ស្លាបព្រាក្នុងទឹក 250 មីលីលីត្រ។ ផឹក 1 tsp ។ រៀងរាល់ ១០ នាទីម្តង។
- យកការលាបថ្នាំផ្សំពីរុក្ខជាតិ។
- ដើម្បីពន្លឿនការលុបបំបាត់ជាតិពុលចេញពីរាងកាយបរិភោគបន្តិចម្តង ៗ ។ វាជាការប្រសើរក្នុងការញ៉ាំតែនំកែកឃឺ។
ការព្យាបាលអាស្រ័យលើលក្ខណៈបុគ្គលនៃរាងកាយ។ វាត្រូវតែត្រូវបានគ្រប់គ្រងយ៉ាងតឹងរ៉ឹងដោយគ្រូពេទ្យដែលចូលរួម។
៧៦. កូឡេស្តេរ៉ុល។ វិធីនៃការចូលការប្រើប្រាស់និងការដកខ្លួនចេញពីរាងកាយ។ កូឡេស្តេរ៉ុល។ ជីវគីមីជីវគីមីកូឡេស្តេរ៉ុលដំណាក់កាលរបស់វា។ បទប្បញ្ញត្តិនៃការសំយោគ។
កូលេស្តេរ៉ុលគឺជាស្តេរ៉ូអ៊ីតជាក់លាក់ចំពោះសារពាង្គកាយសត្វ។ វាត្រូវបានសំយោគនៅក្នុងជាលិការរបស់មនុស្សជាច្រើនប៉ុន្តែកន្លែងសំយោគសំខាន់គឺថ្លើម។ នៅក្នុងថ្លើមច្រើនជាង 50% នៃកូលេស្តេរ៉ុលត្រូវបានគេសំយោគនៅក្នុងពោះវៀនតូច - 15-20% ចំណែកកូលេស្តេរ៉ុលដែលនៅសល់ត្រូវបានគេសំយោគនៅក្នុងស្បែកក្រពេញអ័រឌែននិងហ្គូណាដ។ កូលេស្តេរ៉ុលប្រហែល ១ ក្រាមត្រូវបានគេសំយោគក្នុងមួយថ្ងៃនៅក្នុងខ្លួន ៣០០-៥០០ មីលីក្រាមត្រូវបានលេបចូលជាមួយអាហារ (រូបភាព ៨-៦៥) ។ កូលេស្តេរ៉ុលអនុវត្តមុខងារជាច្រើន: វាគឺជាផ្នែកមួយនៃភ្នាសកោសិកាទាំងអស់ហើយជះឥទ្ធិពលដល់លក្ខណៈសម្បត្តិរបស់វាដើរតួជាស្រទាប់ខាងក្រោមដំបូងក្នុងការសំយោគអាស៊ីតទឹកប្រមាត់និងអរម៉ូនស្តេរ៉ូអ៊ីត។ ភ្នាក់ងារមុននៅក្នុងផ្លូវមេតាប៉ូលីសនៃការសំយោគកូលេស្តេរ៉ុលក៏ប្រែទៅជា ubiquinone ដែលជាសមាសធាតុនៃសង្វាក់ដង្ហើមនិង dolichol ដែលត្រូវបានចូលរួមក្នុងការសំយោគ glycoproteins ។ ដោយសារតែក្រុមអ៊ីដ្រូខ្យូមរបស់ខ្លួនកូលេស្តេរ៉ុលអាចបង្កើតជាអេស្ត្រូសែនជាមួយនឹងអាស៊ីតខ្លាញ់។ កូលេស្តេរ៉ុលអេទីតមានលើសនៅក្នុងឈាមហើយត្រូវបានរក្សាទុកក្នុងបរិមាណតិចតួចក្នុងប្រភេទកោសិកាមួយចំនួនដែលប្រើវាជាស្រទាប់ខាងក្រោមសម្រាប់សំយោគសារធាតុផ្សេងៗ។ កូលេស្តេរ៉ុលនិងអេស្ត្រូសែនរបស់វាគឺជាម៉ូលេគុលអ៊ីដ្រូហ្វីបដូច្នេះពួកវាត្រូវបានដឹកដោយឈាមដែលជាផ្នែកមួយនៃប្រភេទថ្នាំផ្សេងៗគ្នា។ ការផ្លាស់ប្តូរកូលេស្តេរ៉ុលគឺស្មុគស្មាញបំផុត - សម្រាប់តែការសំយោគរបស់វាប៉ុណ្ណោះប្រតិកម្មប្រហែល ១០០ ជាប់គ្នាគឺចាំបាច់។ សរុបទៅមានប្រូតេអ៊ីនប្រមាណ ៣០០ ផ្សេងៗគ្នាចូលរួមក្នុងការរំលាយអាហារកូលេស្តេរ៉ុល។ ភាពមិនចុះសម្រុងនៃការរំលាយអាហារកូលេស្តេរ៉ុលនាំឱ្យមានជំងឺមួយក្នុងចំណោមជំងឺទូទៅបំផុត - ជំងឺក្រិនសរសៃឈាម។ អត្រាមរណៈភាពពីផលប៉ះពាល់នៃជំងឺបេះដូង (ការរំលោភបំពានលើសរសៃឈាមខួរក្បាល) នាំអោយមានរចនាសម្ព័ន្ធទាំងមូលនៃមរណភាព។ ជំងឺអ៊ប៉សសឺឡែនគឺជា "ជំងឺប៉ូលីនជីន" ពោលគឺអាយ។ មានកត្តាជាច្រើនចូលរួមនៅក្នុងការអភិវឌ្ឍន៍របស់វាដែលសំខាន់បំផុតគឺតំណពូជ។ ការប្រមូលផ្តុំកូលេស្តេរ៉ុលក្នុងរាងកាយនាំឱ្យមានការវិវត្តនៃជំងឺទូទៅមួយទៀត - ជំងឺគ្រួសក្នុងប្រមាត់។
A. សំយោគកូលេស្តេរ៉ុលនិងបទបញ្ជារបស់វា
ប្រតិកម្មសំយោគកូឡេស្តេរ៉ុលកើតឡើងនៅក្នុងស៊ីតូកូលនៃកោសិកា។ នេះគឺជាផ្លូវមេតាប៉ូលីសដ៏វែងបំផុតមួយនៅក្នុងខ្លួនមនុស្ស។
ផេនីឡីកុនណូរីយ៉ា

ផេនីឡីកុនណូរីយ៉ា - ការបំពានតំណពូជនៃការរំលាយអាហារអាស៊ីដអាមីណូដោយសារតែកង្វះអង់ស៊ីមថ្លើមដែលពាក់ព័ន្ធនឹងការរំលាយអាហាររបស់ phenylalanine ទៅ tyrosine ។ គស្ញដំបូងនៃជំងឺ phenylketonuria គឺក្អួតល្ហិតល្ហៃឬផ្ចង់អារម្មណ៍ខ្លាំងក្លិនផ្សិតពីទឹកនោមនិងស្បែកការវិវឌ្ឍន៍ផ្នែកចិត្តសាស្ត្រយឺតយ៉ាវមានរោគសញ្ញាយឺតយ៉ាវធម្មតារួមមានអូហ្គីហ្វីលីនការពន្យាពេលនៃការវិវឌ្ឍន៍នៃរាងកាយការប្រកាច់ការផ្លាស់ប្តូរស្បែកអេកូស័រជាដើម។ ការធ្វើរោគវិនិច្ឆ័យជាបន្តបន្ទាប់រួមមានការធ្វើតេស្តហ្សែនម៉ូលេគុលការប្តេជ្ញាចិត្តនៃការផ្តោតអារម្មណ៍របស់ phenylalanine ក្នុងឈាមការវិភាគជីវគីមីទឹកនោម EEG និង MRI នៃខួរក្បាល។ ការព្យាបាលជំងឺ phenylketonuria គឺត្រូវធ្វើតាមរបបអាហារពិសេស។

ព័ត៌មានទូទៅ
Phenylketonuria (ជំងឺរបស់ Felling, phenylpyruvic oligophrenia) គឺជារោគសាស្ត្រដែលកំណត់ដោយហ្សែនពីកំណើតដែលត្រូវបានកំណត់លក្ខណៈដោយការថយចុះជាតិអ៊ីដ្រូឡាលីនហ្វីណីឡាទីនការចុះខ្សោយនៃអាស៊ីតអាមីណូនិងការរំលាយរបស់វានៅក្នុងអង្គធាតុរាវនិងជាលិកាខាងសរីរវិទ្យាបន្ទាប់មកខូចខាតយ៉ាងធ្ងន់ធ្ងរដល់ប្រព័ន្ធសរសៃប្រសាទកណ្តាល។ រោគមុនត្រូវបានពិពណ៌នាដោយ A. Felling ដំបូងក្នុងឆ្នាំ ១៩៣៤ វាកើតឡើងជាមួយប្រេកង់ ១ ករណីចំពោះទារកទើបនឹងកើត ១០.០០០ នាក់។ នៅក្នុងរយៈពេលនៃទារកទើបនឹងកើត, phenylketonuria មិនមានការបង្ហាញរោគសញ្ញាទេទោះជាយ៉ាងណាក៏ដោយការទទួលទានថ្នាំ phenylalanine ជាមួយអាហារបណ្តាលឱ្យមានការលេចឡើងនៃជំងឺនេះនៅពាក់កណ្តាលដំបូងនៃជីវិតហើយជាបន្តបន្ទាប់នាំឱ្យមានការវិវត្តធ្ងន់ធ្ងរនៃកុមារ។ នោះហើយជាមូលហេតុដែលការរកឃើញរោគសញ្ញាមុននៃរោគ phenylketonuria ចំពោះទារកទើបនឹងកើតគឺជាភារកិច្ចសំខាន់បំផុតនៃរោគសរសៃប្រសាទផ្នែករោគកុមារនិងហ្សែន។
មូលហេតុនៃជំងឺ Phenylketonuria
Phenylketonuria គឺជាជំងឺមរតកមួយដែលមានការឈប់សំរាកដោយស្វ័យប្រវត្តិ។ នេះមានន័យថាសម្រាប់ការវិវឌ្ឍន៍នៃរោគសញ្ញាគ្លីនិកនៃ phenylketonuria កុមារត្រូវទទួលមរតកហ្សែនមួយច្បាប់ពីឪពុកម្តាយទាំងពីរដែលជាអ្នកផ្ទុកហ្សែនហ្សែនហ្សែនហ្សែន។
ភាគច្រើនការវិវឌ្ឍន៍នៃ phenylketonuria គឺបណ្តាលមកពីការផ្លាស់ប្តូរហ្សែនក្នុងការបំលែងហ្សែនអង់ស៊ីម phenylalanine-4-hydroxylase ដែលមានទីតាំងស្ថិតនៅលើដៃវែងនៃក្រូម៉ូសូមទី ១២ (ទីតាំង ១២q២២-q២៤.១) ។ នេះគឺជាប្រភេទបុរាណដែលគេហៅថា phenylketonuria ដែលមានចំនួន ៩៨% នៃករណីទាំងអស់នៃជំងឺនេះ។ hyperphenylalaninemia អាចឈានដល់ ៣០ មីលីក្រាមនិងខ្ពស់ជាងនេះ។ ប្រសិនបើមិនបានព្យាបាលទេវ៉ារ្យ៉ង់ហ្វីលីពុនណូរីយ៉ានេះត្រូវបានអមដោយការវិកលចរិកជ្រៅ។
បន្ថែមពីលើទម្រង់បុរាណការព្យាបាលដោយប្រើថ្នាំ atypical phenylketonuria ត្រូវបានសម្គាល់ដោយបន្តរោគសញ្ញារោគសញ្ញាដូចគ្នាប៉ុន្តែមិនមានលក្ខណៈត្រឹមត្រូវទេក្នុងការកែតម្រូវដោយការព្យាបាលដោយរបបអាហារ។ ទាំងនេះរួមមានប្រភេទ phenylketonuria ប្រភេទទី ២ (កង្វះជាតិទឹក dehydroterterin reductase), ប្រភេទ phenylketonuria ប្រភេទទី ៣ (កង្វះតេតារ៉ាឌីរ៉ូប៊ីប៉ូទីន) និងប្រភេទផ្សេងៗទៀតដែលកម្រមាន។
ប្រូបាប៊ីលីតេនៃការផ្តល់កំណើតដល់កុមារដែលមាន phenylketonuria កើនឡើងជាមួយនឹងអាពាហ៍ពិពាហ៍ជិតស្និទ្ធ។
រោគសាស្ត្រនៃជំងឺ phenylketonuria
ទម្រង់បុរាណនៃ phenylketonuria គឺផ្អែកលើភាពមិនគ្រប់គ្រាន់នៃអង់ស៊ីម phenylalanine-4-hydroxylase ដែលពាក់ព័ន្ធនឹងការបំលែង phenylalanine ទៅ tyrosine ក្នុង hepatocyte mitochondria ។ នៅក្នុងវេន, tyramine ដេរីវេទីរ៉ូអ៊ីតគឺជាសម្ភារៈចាប់ផ្តើមសម្រាប់ការសំយោគ catecholamines (adrenaline និង norepinephrine) និង diiodotyrosine សម្រាប់ការបង្កើត thyroxine ។ លើសពីនេះទៀតការបង្កើតសារធាតុពណ៌មេឡាញីនគឺជាលទ្ធផលនៃការរំលាយអាហារ phenylalanine ។
កង្វះតំណពូជនៃអង់ស៊ីម phenylalayin-4-hydroxylase នៅក្នុង phenylketonuria នាំឱ្យមានការរំលោភលើការកត់សុីនៃ phenylalanine ពីអាហារដែលបណ្តាលឱ្យកំហាប់របស់វានៅក្នុងឈាម (phenylalaninemia) និងសារធាតុរាវ cerebrospinal មានការកើនឡើងគួរឱ្យកត់សម្គាល់ហើយកម្រិតនៃ tyrosine មានការថយចុះ។ លើសពី phenylalanine ត្រូវបានលុបចោលដោយការកើនឡើងនៃការបញ្ចេញទឹកនោមរបស់មេតាប៉ូលីសរបស់វា - អាស៊ីត phenylpyruvic, phenylmilactic និង phenylacetic ។
ការរំខាននៃការរំលាយអាហារអាស៊ីតអាមីណូត្រូវបានអមដោយការថយចុះការខ្សោយនៃសរសៃប្រសាទដែលជាការថយចុះនៃការបង្កើតកោសិកាសរសៃប្រសាទ (ឌីប៉ូតាមីនសឺរ៉ូទីនជាដើម) ដែលបង្កឱ្យមានយន្តការបង្កជំងឺនៃការវិវត្តនៃខួរក្បាលនិងជំងឺវង្វេងវង្វេង។
រោគសញ្ញានៃ phenylketonuria
ទារកទើបនឹងកើតដែលមាន phenylketonuria មិនមានរោគសញ្ញារោគសញ្ញានៃជំងឺនេះទេ។ ជាធម្មតាការបង្ហាញនៃ phenylketonuria ចំពោះកុមារកើតឡើងនៅអាយុ 2-6 ខែ។ ជាមួយនឹងការចាប់ផ្តើមនៃការបំបៅប្រូតេអ៊ីននៃទឹកដោះម្តាយឬជំនួសរបស់វាចាប់ផ្តើមចូលទៅក្នុងខ្លួនទារកដែលនាំឱ្យមានការវិវត្តនៃរោគសញ្ញាដំបូងដែលមិនជាក់លាក់ - សន្លឹមពេលខ្លះការថប់បារម្ភនិងភាពរំភើបហួសហេតុការកន្ត្រាក់សាច់ដុំជំងឺវង្វេងវង្វាន់រោគសញ្ញាអាការជម្ងឺ។ សញ្ញាមួយនៃរោគសញ្ញារោគសាស្ត្រដំបូងនៃជំងឺ phenylketonuria គឺការក្អួតចង្អោរដែលជារឿយៗត្រូវបានគេយល់ច្រឡំថាជាការបង្ហាញនៃជំងឺក្រិនសរសៃឈាម។
នៅពាក់កណ្តាលទីពីរនៃឆ្នាំនេះភាពយឺតយ៉ាវរបស់កុមារក្នុងការអភិវឌ្ឍផ្នែកចិត្តវិទ្យាក្លាយជាការកត់សម្គាល់។ ក្មេងក្លាយទៅជាមិនសូវសកម្មព្រងើយកណ្តើយឈប់ទទួលស្គាល់មនុស្សជាទីស្រឡាញ់កុំព្យាយាមអង្គុយហើយឈរជើងរបស់គាត់។ សមាសធាតុទឹកនោមនិងញើសមិនធម្មតាបណ្តាលឱ្យមានក្លិនលក្ខណៈ“ កណ្តុរ” (ក្លិនរបស់ផ្សិត) ចេញពីរាងកាយ។ ជារឿយៗមានការរមាស់នៃស្បែក, ជំងឺរលាកស្បែក, ជំងឺត្រអក, ជំងឺក្រិន។
ចំពោះកុមារដែលមានសារធាតុ phenylketonuria ដែលមិនទទួលការព្យាបាល, មីក្រូជីហ្វីល, រោគវិទ្យា, ក្រោយមក (ក្រោយរយៈពេល 1,5 ឆ្នាំ) teething, hypoplasia enamel ត្រូវបានរកឃើញ។ ការពន្យាពេលក្នុងការអភិវឌ្ឍការនិយាយត្រូវបានគេកត់សំគាល់ហើយដោយរយៈពេល ៣-៤ ឆ្នាំនៃអូលីហ្វៀរៀយ៉ាងជ្រៅនិងអវត្តមានស្ទើរតែទាំងស្រុងនៃការនិយាយត្រូវបានរកឃើញ។
កុមារដែលមានជំងឺ phenylketonuria មានកាយវិការអសមត្ថភាពដែលជាញឹកញាប់មានបញ្ហាបេះដូងពីកំណើត, ការចុះខ្សោយនៃស្វយ័ត (បែកញើស, Acrocyanosis, សរសៃឈាមអារទែ) និងទទួលរងពីការទល់លាមក។ លក្ខណៈ Phenotypic របស់កុមារដែលទទួលរងពី phenylketonuria រួមមានស្បែកស្រាលភ្នែកនិងសក់។ កុមារដែលមានសារធាតុ phenylketonuria ត្រូវបានកំណត់លក្ខណៈដោយអ្នកកាត់ដេរជាក់លាក់មួយ (អវយវៈខាងលើនិងខាងក្រោមកោងនៅសន្លាក់) ញ័រដៃស្វាហាប់ដកដង្ហើមញាប់ញ័រនិង hyperkinesis ។
ការបង្ហាញគ្លីនិកនៃប្រភេទទី ២ phenylketonuria ត្រូវបានកំណត់ដោយការថយចុះស្មារតីធ្ងន់ធ្ងរ, ឆាប់ខឹង, កើនឡើង, ប្រកាច់, រមួលក្រពើនិង hyperreflexia សរសៃពួរ។ ការវិវត្តនៃជំងឺនេះអាចនាំឱ្យមានការស្លាប់របស់កុមារដែលមានអាយុពី 2 ទៅ 3 ឆ្នាំ។
នៅពេលដែលសារធាតុ phenylketonuri ប្រភេទ III មានការរីកចម្រើននៃគស្ញ: microcephaly, oligophrenia, spet tetraparesis ។
ការធ្វើរោគវិនិច្ឆ័យរោគ phenylketonuria
បច្ចុប្បន្ននេះការធ្វើរោគវិនិច្ឆ័យរោគ phenylketonuria (ក៏ដូចជាជម្ងឺ galactosemia, ជំងឺក្រពេញទីរ៉ូអ៊ីតពីកំណើត, រោគសញ្ញា adrenogenital និងជំងឺ cystic fibrosis) គឺជាផ្នែកមួយនៃកម្មវិធីពិនិត្យលើទារកទើបនឹងកើត។
ការធ្វើតេស្តពិនិត្យត្រូវបានធ្វើឡើងក្នុងរយៈពេល ៣-៥ ថ្ងៃនៃជីវិតពេញមួយជីវិតនិងរយៈពេល ៧ ថ្ងៃនៃទារកដែលកើតមិនគ្រប់ខែដោយយកសំណាកឈាម capillary លើទម្រង់ក្រដាសពិសេស។ ប្រសិនបើរកឃើញ hyperphenylalanemia ច្រើនជាង ២,២ មីលីក្រាមនៃកុមារត្រូវបានបញ្ជូនទៅរកហ្សែនកុមារដើម្បីធ្វើការពិនិត្យម្តងទៀត។
ដើម្បីបញ្ជាក់ពីការធ្វើរោគវិនិច្ឆ័យនៃ phenylketonuria, កំហាប់នៃ phenylalanine និង tyrosine ក្នុងឈាមត្រូវបានពិនិត្យសកម្មភាពរបស់អង់ស៊ីមជំងឺទឹកនោមផ្អែម (phenylalanine hydroxylase) ត្រូវបានកំណត់, ការពិនិត្យជីវគីមីនៃទឹកនោម (ការប្តេជ្ញាចិត្តនៃអាស៊ីត ketonic), ការរំលាយអាហាររបស់ catecholamines ក្នុងទឹកនោម។ ល។
ពិការភាពហ្សែននៅក្នុង phenylketonuria អាចត្រូវបានគេរកឃើញសូម្បីតែក្នុងកំឡុងពេលមានផ្ទៃពោះក្នុងកំឡុងពេលធ្វើរោគវិនិច្ឆ័យរាតត្បាតមុនពេលមានផ្ទៃពោះរបស់ទារក (chorionbiopsy, amniocentesis, cordocentesis) ។
ការធ្វើរោគវិនិច្ឆ័យឌីផេរ៉ង់ស្យែល phenylketonuria ត្រូវបានអនុវត្តជាមួយនឹងការប៉ះទង្គិចពីកំណើតចំពោះទារកទើបនឹងកើត, ការឆ្លងមេរោគក្នុងពោះវៀននិងជំងឺមេតាប៉ូលីសដទៃទៀត។
ការព្យាបាលដោយប្រើថ្នាំ Phenylketonuria
កត្តាមូលដ្ឋានក្នុងការព្យាបាលជម្ងឺ phenylketonuria គឺជារបបអាហារដែលកំណត់ការទទួលទានប្រូតេអ៊ីននៅក្នុងខ្លួន។ ការព្យាបាលត្រូវបានណែនាំឱ្យចាប់ផ្តើមនៅកំហាប់ phenylalanine> ៦ មីលីក្រាម។ ល្បាយពិសេសត្រូវបានបង្កើតឡើងសម្រាប់ទារក - Afenilak, Lofenilak សម្រាប់កុមារដែលមានអាយុលើសពី ១ ឆ្នាំ - Tetrafen, មិនមានជាតិ Phenyl, មានអាយុលើសពី ៨ ឆ្នាំ - Maxamum-XP និងផ្សេងៗទៀត។ មូលដ្ឋានគ្រឹះនៃរបបអាហារគឺអាហារប្រូតេអ៊ីនតិច - ផ្លែឈើបន្លែទឹកផ្លែឈើអ៊ីដ្រូអ៊ីដ្រាតនិងល្បាយអាស៊ីដអាមីណូ។ ។ ការពង្រីករបបអាហារគឺអាចធ្វើទៅបានបន្ទាប់ពីរយៈពេល 18 ឆ្នាំទាក់ទងនឹងការកើនឡើងនៃភាពអត់ធ្មត់ចំពោះថ្នាំ phenylalanine ។ អនុលោមតាមច្បាប់របស់ប្រទេសរុស្ស៊ីការផ្តល់អាហាររូបត្ថម្ភវេជ្ជសាស្រ្តសម្រាប់មនុស្សដែលទទួលរងពី phenylketonuria គួរតែមិនគិតថ្លៃ។
អ្នកជំងឺត្រូវបានគេចេញវេជ្ជបញ្ជាឱ្យទទួលទានសារធាតុរ៉ែវីតាមីននៃក្រុមខ។ ល។ នេះបើយោងតាមការចង្អុលបង្ហាញ - ថ្នាំ nootropic, anticonvulsants ។ នៅក្នុងការព្យាបាលដោយស្មុគស្មាញនៃថ្នាំ phenylketonuria ការម៉ាស្សាទូទៅការព្យាបាលដោយការធ្វើលំហាត់ប្រាណនិងការចាក់ម្ជុលវិទ្យាសាស្ត្រត្រូវបានគេប្រើយ៉ាងទូលំទូលាយ។
កុមារដែលទទួលរងពីរោគ phenylketonuria ស្ថិតនៅក្រោមការត្រួតពិនិត្យរបស់គ្រូពេទ្យកុមារនិងគ្រូពេទ្យជំនាញខាងសរសៃប្រសាទក្នុងតំបន់ហើយជារឿយៗត្រូវការជំនួយពីអ្នកព្យាបាលការនិយាយនិងអ្នកឯកទេសខាងរោគសាស្ត្រ។ ការត្រួតពិនិត្យដោយយកចិត្តទុកដាក់លើស្ថានភាព neuropsychic របស់កុមារការត្រួតពិនិត្យកម្រិតនៃ phenylalanine នៅក្នុងសូចនាករឈាមនិង electroencephalogram គឺចាំបាច់។
ទំរង់ Atypical នៃ phenylketonuria ដែលមិនអាចទទួលយកបានក្នុងការព្យាបាលរបបអាហារតម្រូវឱ្យមានការតែងតាំងថ្នាំ hepatoprotectors, anticonvulsants, ការព្យាបាលជំនួសដោយ levodopa, 5-hydroxytryptophan ។
ការព្យាករណ៍និងការការពារជំងឺ phenylketonuria
ធ្វើការពិនិត្យម៉ាស់ហ្វីណីលីនតុនៀនៅដំណាក់កាលទារកទើបនឹងកើតអនុញ្ញាតឱ្យអ្នករៀបចំការព្យាបាលរបបអាហារមុននិងការពារការខូចខាតខួរក្បាលធ្ងន់ធ្ងរមុខងារថ្លើមចុះខ្សោយ។ ជាមួយនឹងការតែងតាំងដំបូងនៃរបបអាហារបំបាត់សម្រាប់ថ្នាំ phenylketonuria បុរាណការព្យាករណ៍សម្រាប់ការអភិវឌ្ឍកុមារគឺល្អ។ ជាមួយនឹងការព្យាបាលយឺតការព្យាករណ៍សម្រាប់ការអភិវឌ្ឍផ្លូវចិត្តគឺខ្សោយ។
ការបងា្ករផលវិបាកនៃថ្នាំ phenylketonuria មាននៅក្នុងការពិនិត្យរកមើលទារកទើបនឹងកើតការចេញវេជ្ជបញ្ជាដំបូងនិងការអនុលោមតាមពេលវេលាយូរជាមួយអាហារបំប៉ន។
ដើម្បីវាយតម្លៃពីហានិភ័យនៃការផ្តល់កំណើតដល់កុមារដែលមានសារធាតុ phenylketonuria ការប្រឹក្សាពីហ្សែនបឋមគួរតែត្រូវបានផ្តល់ឱ្យគូស្វាមីភរិយាដែលមានកូនឈឺរួចហើយមានទំនាក់ទំនងស្នេហានិងមានសាច់ញាតិដែលមានជំងឺនេះ។ ស្ត្រីដែលមានជាតិ phenylketonuria ដែលកំពុងរៀបចំផែនការមានផ្ទៃពោះគួរតែធ្វើតាមរបបអាហារដ៏តឹងរឹងមុនពេលមានផ្ទៃពោះនិងអំឡុងពេលមានផ្ទៃពោះដើម្បីមិនរាប់បញ្ចូលការកើនឡើងនៃកម្រិត phenylalanine និងការរំលាយអាហាររបស់វានិងការថយចុះការលូតលាស់របស់ទារកដែលមានសុខភាពហ្សែន។ ហានិភ័យនៃការមានកូនដែលមាន phenylketonuria ចំពោះឪពុកម្តាយដែលផ្ទុកហ្សែនដែលមានជម្ងឺគឺ 1: 4 ។